Научный коллектив под руководством профессора Ван Ядуна с факультета вычислительной техники Харбинского политехнического университета (ХПУ) достиг значительного прогресса в разработке алгоритмов биоинформатики, предложив универсальный метод определения клеточного состава на основе мультиомиксных данных. Этот метод ознаменовал переход от «отдельных инструментов» к «универсальной платформе» и установил новый стандарт для комплексного мультиомиксного анализа.
Результаты исследования под заголовком «DECODE: deep learning-based common deconvolution framework for various omics data» опубликованы в журнале Nature Methods. В том же номере журнала в рубрике «Новости и мнения» (News and Views) была опубликована специальная статья, подготовленная по приглашению редакции командой под руководством члена Австралийской академии наук профессора Робина Гасссера, в которой были подробно прокомментированы результаты исследования.
Типы клеток, входящих в состав ткани, и их динамические состояния играют критическую роль в понимании сложных биологических систем и механизмов возникновения и развития заболеваний. Омиксные данные, получаемые из образцов тканей, обычно представляют собой смешанные сигналы от различных типов клеток. Поэтому для определения их пропорций необходимо использовать алгоритмы деконволюции (разделения сигналов). В последние годы с развитием таких мультиомиксных технологий, как транскриптомика, протеомика и метаболомика, происходит накопление данных крупномасштабных когортных исследований. Однако существующие алгоритмы деконволюции, как правило, разработаны для одного типа омиксных данных и с трудом поддаются унификации для анализа данных разных омиксных платформ, что в значительной степени ограничивает комплексное использование мультиомиксных данных.
Для решения этой проблемы команда профессора Ван Ядуна разработала универсальный алгоритм DECODE. Данный метод может быть одинаково применён к данным транскриптомики, протеомики и метаболомики, а также позволяет осуществлять совместный анализ типов клеток и их состояний. Это восполняет многолетний пробел в области деконволюции метаболомических данных – отсутствие универсальной алгоритмической платформы.
Исследовательская группа разработала единый алгоритм, состоящий из нескольких модулей. Сначала генерируются симулированные данные для обучения; затем используется переносное состязательное обучение для выравнивания признаков, что эффективно устраняет батч-эффекты между различными платформами и наборами данных. Далее, с помощью комбинации контрастного обучения и механизмов самовнимания, осуществляется фильтрация шумов для реконструкции истинных клеточных характеристик. Синергическое действие этих модулей позволяет DECODE стабильно восстанавливать информацию о типах клеток и их состояниях даже при наличии сложных данных.
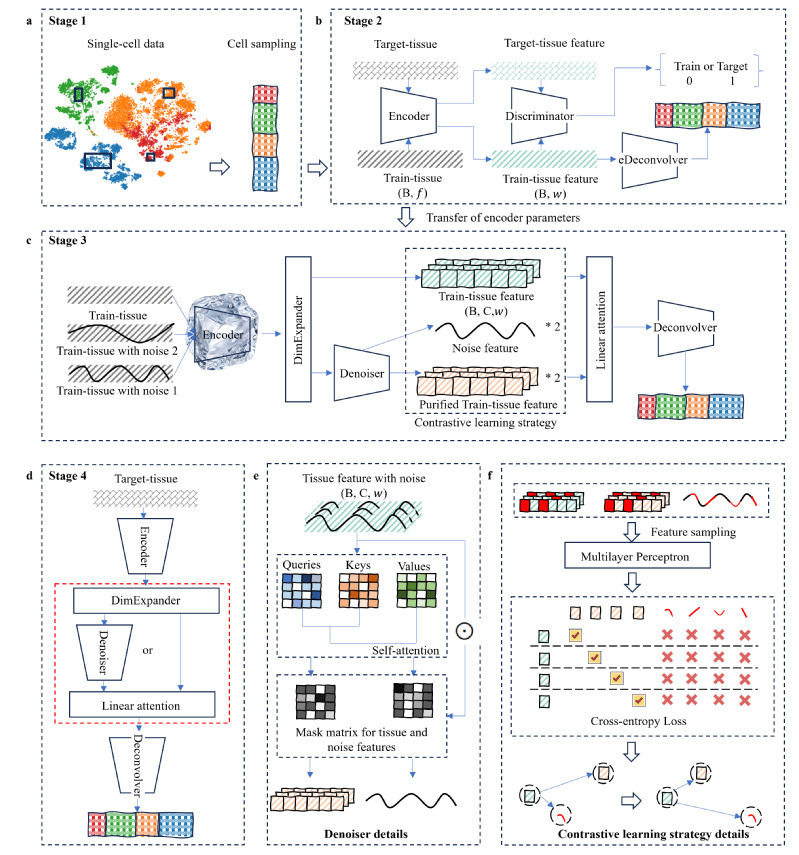
Блок-схема алгоритма DECODE
Результаты системной оценки показывают, что DECODE значительно превосходит основные современные алгоритмы работы с различными типами омиксных данных, состояниями заболеваний, наборами данных и измерительными платформами. Более того, метод сохраняет высокую точность определения клеточного состава даже при неполных референсных данных на уровне отдельных клеток, что демонстрирует его исключительную обобщающую способность и робастность.
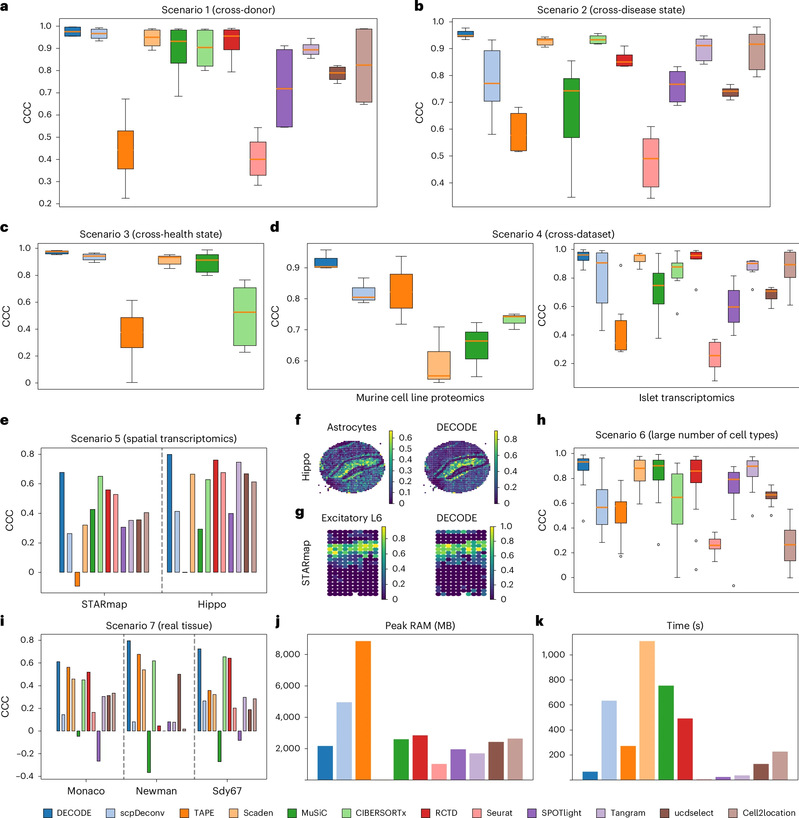
Экспериментальные результаты DECODE на различных типах омиксных данных, состояниях заболеваний, наборах данных и измерительных платформах
При анализе мультиомиксных данных когортного анализа по раку молочной железы DECODE выявил важные изменения в составе иммунных клеток в процессе прогрессирования опухоли. Например, на стадии неметастатических предраковых изменений наблюдалось значительное увеличение доли T-клеток и заметное снижение доли B-клеток. Это открытие согласуется с выводами предыдущих исследований о том, что инфильтрация T-клеток тесно связана с благоприятным прогнозом. Дальнейший анализ субпопуляций показал, что на ранних стадиях заболевания происходит увеличение доли CD4⁺T-клеток, CD8⁺ T-клеток и пролиферирующих T-клеток, в то время как увеличение доли B-клеток на поздних стадиях рака происходит в основном за счёт наивных B-клеток. Это может отражать нарушение иммунной функции опухоли и усиление её метастатического потенциала.
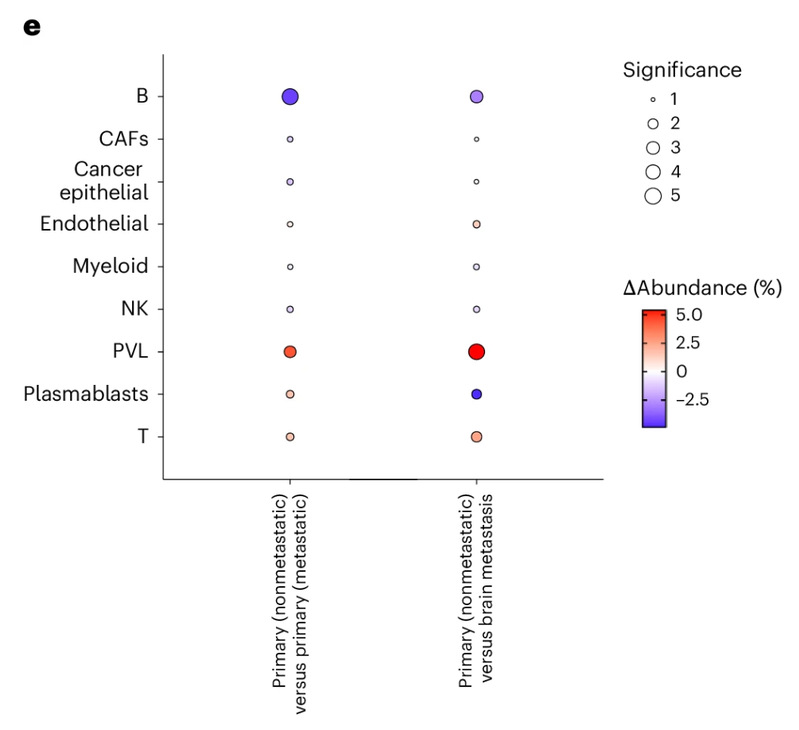
Результаты применения метода DECODE к данным когортного анализа рака молочной железы
Харбинский политехнический университет является первой аффилиацией авторов. Профессор Ван Ядун является автором-корреспондентом публикации. Профессор Чжао Тяньи Факультета естественных наук и медицины, аспиранты Лю Жэньцзе и Сунь Юйчжи Факультета вычислительной техники являются первыми авторами. Исследование поддержано грантом Национального фонда естественных наук Китая и другими источниками финансирования.
Ссылка на статью: https://www.nature.com/articles/s41592-026-03007-y